Thermal Decomposition of Solid Energetic Materials
Y. M. Burov
Institute of Problems of Chemical Physics, Russian Academy of Sciences, 142432 Chernogolovka, Moscow Region , Russian Federation . E-mail: yuburov@icp.ac.ru
Decomposition of the organic compounds in the solid phase occurs much more slowly than in the liquid and gas phases [1-3]. The retardation effect of the crystalline lattice (REL), which can be defined as the ratio of rate constants of decomposition in the liquid phase to that in the solid phase ( k liq /k s ) , is often the only factor that causes the stability of the substance and its suitability for use after prolong storage. This situation is characteristic of medicines, explosives, and initiators of chain processes. Experimental determination of REL and development of methods suitable to predict the properties of new substances are an important aspect of the general theory of the stability of organic compounds. Development in this field is restricted because the date on k s values are limited and the theoretical models of monomolecular reactions in the solid phase cannot be verified.
Solid phase decomposition reactions are usually accompanied by several side processes, such as evaporation and fast decomposition of compounds in the gas phase, melting on admixture and products autocatalysis at the early stages of the process, effects of premelting near points of phase transitions, and others. Due to side reactions, the observed k s values often exceed the true values by one and even two orders of magnitude. The methods for taking into account secondary factors [2] are used to a full extent only for several compounds.
Therefore, the purpose of this work is to obtain, firstly, a sufficiently representative array of correct k s and REL = k liq /k s values and, secondly, the dependence of REL on the physical properties of the crystal and other parameters used in theoretical models.
Experimental
Decomposition rates were measured by the manometric method. The reaction course was monitored at conversions of 0.01 – 1.00%, which allowed one to avoid the influence of autocatalytic processes and topochemical regime of the reaction. To eliminate unstable or catalytic admixtures, the substances were purified by sublimation onto a heated support or y recrystallization from different solvents with thorough drying. The purification was carried until a constant decomposition rate was achieved.
To increase the sensitivity of the method, we used such loadings of the substance that the ratio of the sample weight to the volume of the reaction vessel was 0.5 g cm -3 . Thus, the amount of the substance in the vapor phase was insignificant as compared to the weight of the solid sample, and the reaction in vapor could be neglected even at the highest possible difference in the rates in the gas and solid phases (four orders of magnitude) [4 – 6].
The k s values were calculated from the time of achievement of the degree of decomposition of 0.1%. For calculation of the conversion, the stoichiometric coefficient of gas release determined by the decomposition of the substance in melt was used.
Table 1. Retardation effect of the crystalline lattice and physical properties of the substances
N
Compound
k liq /k s
m . p ., 0 С
.10 10 , P а -1
V 0 , cm 3 /mol
1
[FC(NO 2 ) 2 CH 2 ] 2 NNO 2
4
86
3,530
173,9
2
[F 2 NC(NO 2 ) 2 CH 2 ] 2 NNO 2
4
102
-
198,0
3
C(NO 2 ) 3 C (NO 2 ) 3
6
140
5,500
169,4
4
PhN=N-NHPh
6
101
-
-
5
[C(NO 2 ) 3 CH 2 ] 2 NNO 2
10
95,5
1,050
199,0
6
F 2 NC(NO 2 ) 2 CH 2 NNO 2 (CH 2 ) 2 C (NO 2 ) 2 NF 2
The REL values for 22 compounds calculated using the correct k s values are presented in Table 1. The kinetic data using for composing Table 1 are presented in Table 2. The rate constants obtained for the decomposition in melt (noncatalytic stages) or in inert solvents ware used as k liq . When several data were available, the minimum k s and k liq values taken for the calculation of REL
The REL values ware calculated at temperatures 20 0 C lower then the melting point of the substance when pre-melting effects do not affect the k s value. The melting points of the substances and the volume compressibility ( ) (the parameter characterizing the force of interatomic interaction in the crystal) are also presented in Table 1. The values were calculated by the Rao method [19], and the group increments were borrowed from the literature date [20].
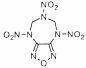
The mechanisms of initial monomolecular stages of decomposition are known for all compounds presented in Table 1: bond cleavage of C-NO 2 ( 2, 3, 5 - 7, 10, 13 ), N-NO 2 ( 1, 14, 16, 17, 19 – 22), O-NO 2 ( 18 ), and N-N ( 4 ) and elimination of N 2 ( 11 ) via formation of the linear transition state or elimination of N 2 ( 9 ) and CO 2 ( 12, 15) molecules through the cyclic transition state.
The majority of reactions are characterized by a positive activation volume, i. e. , they occur with a volume increase in the transition state ( ). For compounds 9 and 11 , one can expect values close to zero, whereas for 12 and 15 , they can even be negative.
It is seen from the data in Table 1 that the REL values vary within very wide limits from 4 to 10 4 . To explain such a broad interval of REL variation, the theory of solid phase reactions should be considered.
Table 2. Kinetic parameters of decomposition of organic compounds in liquid and solid states
Compound
Medium a
T / 0 C
E b
lgA /s -1
k c /s -1
References
1
Solution in TNT
Solid phase
170-210
66
40.0
14.92
1.3·10 -11
3.0·10 -12
4
29
2
Melt
Solid phase
105-120
82
38.5
15.70
3·10 -8
8.8·10 -9
29
29
3
Gas
Solid phase
90-135
120
35.8
16.52
4.0·10 -4
6.6·10 -5
2
29
4
Melt
Solid phase
100-160
80
39.9
17.50
7.2·10 -8
1.2·10 -8
5
29
5
Melt
Solution
Solid phase
110-150
110-165
75
36.8
36.1
15.59
15.06
2.9·10 -8
2.4·10 -8
2.4·10 -9
4
4
29
6
Solution in DNB
Solid phase
100
100
-
-
-
-
1.3·10 -7
1.3·10 -8
29
29
7
Solution in TNT
Solid phase
130-170
100-140
40.3
42.6
16.9
16.4
3·10 -4
1.5·10 -6
29
29
8
Solution in NB
Solid phase
114
114
-
-
-
-
3.3·10 -4
1.3·10 -4
29
29
9
Solution in xylene
Solid phase
70-115
50-100
26.0
28.8
12.10
12.20
1.6·10 -3
5.7·10 -5
6
29
10
Solution in TNT
Solid phase
95
95
-
-
-
-
1.2·10 -3
3.5·10 -5
29
29
11
Solution in DNB
Solid phase
120-180
120-175
36.3
36.7
14.59
12.86
1.4·10 -4
1.6·10 -4
7
29
12
Melt
Solid phase
136-160
115
32.2
-
13.50
-
2.3·10 -5
2.6·10 -7
29
29
13
Solution in DNB
Solid phase
130-180
158
40.7
16.80
2.6·10 -4
2.8·10 -4
4
29
14
Melt
Melt
Melt
Solid phase
140-160
131-155
150-175
80-120
35.2
36.0
35.9
38.9
13.50
13.80
13.50
13.20
2.6·10 -7
1.8·10 -7
1.0·10 -7
1.0·10 -9
8
9
10
2
15
Gas
Solid phase
150-190
120-180
35.2
38.6
13.80
13.40
2.5·10 -4
2.1·10 -6
29
29
16
Melt
Solution in TNB
Solution in NB
Solid phase
135-200
120-260
225-245
185
37.1
37.9
37.1
13.90
14.00
12.00
1.5·10 -8
8.1·10 -8
2.0·10 -6
9.5·10 -9
11
12
12
29
17
Solution in DNB
Solid phase
145-170
150
39.7
15.7
3.3·10 -5
2.3·10 -7
13
29
18
Melt
Solid phase
120-130
40.0
39.0
15.8
12.7
4.7·10 -7
1.3·10 -9
14
2
19
Solution in TNT
Solid phase
170-210
230
38.0
14.5
1·10 -2
2·10 -5
29
29
20
Gas
Solid phase
Solid phase
216
216
204-234
50.1
-
-
18.9
2·10 -3
2.1·10 -5
3.2·10 -4
29
29
15
21
Solution in DNB
Solid phase
160-200
140-190
39.7
39.8
14.3
11.2
1.4·10 -5
1·10 -8
16
17
22
Solution in DNB
Solid phase
171-215
130-180
44.9
37.9
16.0
9.2
3.1·10 -3
3.7·10 -7
16
17
Based on the published date [1], a generalized physical model of monomolecular reactions in the solid phase can be envisaged. A reaction can proceed in the bulk of the crystal or on its surface and on defects of the crystalline lattice. The reaction can proceed in the bulk of the crystal or on its surface and on defects of the crystalline lattice. The reaction occurring on defects has the same activation energy ( E ) as in the liquid phase (or close to it), but the pre-exponential factor includes the coefficient that takes into account the fraction of molecules in disordered sites of the lattice. According to estimates [1, 24, 25] based on the calculation of the number of molecules arranged on the network of dislocations that separate microblocks of the crystal (the linear sizes of the microblocks are 10 -3 – 10 - 5 cm ), the fraction of such reactive molecules is 0.01 – 1.00%. Thus, the reaction on defects limits the k liq /k s ratio to 10 4 , and the reaction in the bulk can be observed only if REL does not exceed 100.
Reaction in the bulk of the crystal requires the formation of a cavity with a volume exceeding the activation volume , so that the leaving group does not experience forces of interatomic attraction. Cavities can be formed by two mechanisms – energetically or entropically.
The macroscopic approach [22, 31 - 33], considering the crystal as an elastic continuous medium, leads to the equation
, (1)
where is the molar volume of the substance, and is the activation volume in the solid phase. This volume is not equal to the value . According to the published data [21], it can be estimated as an increase in the volume of the cell occupied by a molecule due to elongation (by 10 – 15%) of the cleaving bond in the transition state [27], i. e., approximately by 0.2 Å. Extension of the cell by 0.2 Å results in weakening of intermolecular interactions and allows atoms of molecule to converge freely, forming cyclic transition states. Thus, the value depends slightly on the reaction type and on the value and sign of the true activation volume.
The pre-exponential factor of the reaction occurring in the bulk should be the same as that for the reaction in the liquid phase [22]. In [23] added influence of solid state on the pre-exponential factor.
The calculation of REL by Eq. (1), assuming that the cell volume is and its expansion is 0.2 Å, gives values that often coincide by an order of magnitude with the experimental values. For compounds 1, 3, 5, 13, 14, 16, 17, 19, 21, and 22 , i. e., when kinetic data for calculations were available, the calculated REL value was 8, 3, 100, 386, 10, 453, 31, 250, 400, 400, and 3040, respectively. The highest deviations from the experimental values (see Table 1) are observed for molecules with a long chain. In this case, local motions of, not the whole molecule, but only of its fragments containing the reaction center have a substantial effect on the REL values.
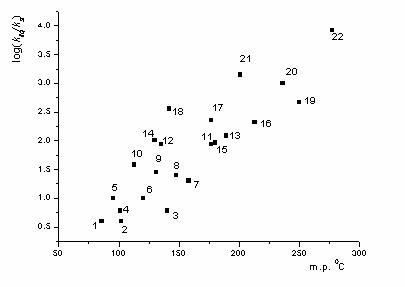
Fig. 1. Dependence REL on volume compressibility.
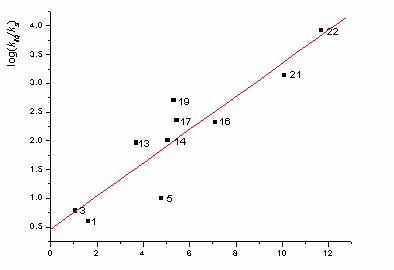
As it's shown in the works [31 - 33], the main assumptions of this theory are contrary to the principles of the thermodynamics. Nevertheless, this theory rather well describes the experiment (see Fig. 1 and 2). Therefore, the new approach - "free volume model" was offered in the works [31 - 33].
At temperatures is higher , where - characteristic temperature, the crystal properties are in the best way described in frameworks cell model. Within the framework of the Lennard – Jones cell model [29], the "free volume" , which one can be computed by a method atom – atom potential [30]
(2)
q - generalized coordinates, - energy of crystal lattice, - interact energy of a molecula with the neighbours in the supposition, that the adjacent moleculas take midpositions, the integrating is carried out on volume basis cells.
Probability of originating of a cavity by the size
Supposing, that [31 – 33]
(3)
the problem of an estimation REL is reducible to finding of a free volume, which one can be computed by Eq. (2), or can be estimated from the degree of thermal expansion
,
- is coefficient of thermal expansion .
The absence of a method of exact estimation of restricts the possibility of using Eq. (1) and (3) for practical calculations, although there is some doubt that considered models adequately reflect the real pattern. Equation (1) is important because it predicts, first, low REL, i.e., the possibility of the reaction occurring purely in the bulk of the sample, and, second, the dependence of REL on the elastic properties of the crystal. Both predictions agree with the experimental data. Only the nonconstant character of the values disturbs the linearity of the dependence of REL on , which is demonstrated by the data in Fig. 1.
The dependence of REL on the melting point of the substance, which is also a good measure of forces of intermolecular interaction, especially for short-chain molecules [28], is presented in Fig. 1, and points for substances with a high molecular weight lie closer to the lower boundary in Fig. 1. For compounds with m.p. < 150 0 C , the REL values are mainly lower than 100 and, hence, we may consider that the reaction in the volume predominates in this case as well.
For the substances with higher melting points, the reaction in the volume is changed by the decomposition on crystal defects, which, as shown above, can limit REL by the interval of 10 2 – 10 4 , which is experimentally observed. Compound 3 (it is not shown in Fig. 1), which represents plastic crystals, falls out of the general dependence. For long-chain compounds 6 and 7 , according to the absolute REL value, decomposition in the bulk is also possible, despite sufficiently high melting points.
Fig. 2. . Dependence REL on melting point.
The experimentally determined value is low and does not usually exceed the measurement error, which is 3 – 4 kcal/mol. Therefore, it is difficult to use this value to estimate REL at different temperatures and to separate volume and local reactions. The latter can be performed using absolute values of REL or melting points of the substances. The experimental data in Table 1 agree with considered models and suggest that for organic compounds with m. p. 100 0 C the irreversible monomolecular reaction occurs in the ideal part of the crystal lattice with REL < 10, and at m. p. > 200 0 C the reaction proceeds on defects only. Within the interval m. p. = 150 – 200 0 C , REL is equal to 10 2 – 10 3 , and the indicated reactions compete with each other. These conclusions give a clear semi-quantitative pattern of changing REL; however, they need further verification. It is necessary to obtain new data on , and REL, including those for reaction with a negative activation volume, as well as to identify directly localized reactions and to compare the decomposition rate with the defectiveness of crystal.
References
• Chemistry of Solid State, Ed. W. E. Garner, Butterworths, London, 1955.
• Yu. M. Burov, G. B. Manelis, and G. M. Nasin, Dokl. Akad. Nauk SSSRR, 1984, 279 , 1142 [Dokl. Chem., 1984 (Engl. Transl.)]
• O. N. Karpukhin, T. V. Pokholok, and V. Ya. Shlyapintokh, Vysokomol. Sedin. A, 1971, 13 , (Engl. Transl.)
• B. L. Korsunski, V. G. Matveev, L. D. Nazina, and G. M. Nazin, 29 th Intern. Annual Conference of ICT (June 3- - July 3, 1998), Karlsruhe, 1998, 60.
• N. Okawara, H. Jamazaki, and E. Imato, J Chem. Soc. Jpn., Ind. Chem. Sect., 1955, 58 , 991.
• B. L. Korsunski, T. A. Apina, and F. I. Dubovitskii, Izv. Akad. Nauk SSSR, Ser. Khim., 1970, 2080 [Bull. Acad. Sci. USSR, Div. Chem. Sci., 1971, 20 (Engl. Transl.)].
• Yu. M. Burov and G. M. Nazin, 29 th Intern. Annual Conference of ICT (June 3- - July 3, 1998), Karlsruhe, 1998, 129.
• F. I. Dubovitskii, G. B. Manelis, and L. P. Smirnov, Zh. Fiz. Khim., 1961, 35 , 521 [J. Phys. Chem. USSR, 1961, 35 (Engl. Transl.)].
• F. I. Dubovitskii, Yu. I Rubtsov, and G. B. Manelis, Izv. Akad. Nauk SSSR, Otd. Khim. Nauk [Bull. Acad. Sci. USSR, Div. Chem. Sci.], 1960, 1763 (in Russian).
• R. S. Stepanov, V. N. Shan'ko, I. P. Medvetskaya, and V. m. Gorodetskaya, in Khimicheskaya fisika protsessov goreniya i vsryva. Kinetika khimicheskikh reaktsii [Chemical Physics of Combustion and Explosion. Kinetics of Chemical Reaction], OIKhF AN SSSR, Chernogolovka, 1977, 56, (in Russian).
• G. V. Sitonina, B. l. Korsunskii, N.F. Pyatakov, V. G. Shvaiko, I. Sh. Abdrakhmanov, and F. I. Dubovtskii , Izv. Akad. Nauk SSSR, Ser. Khim., 1979, 311 [Bull. Acad. Sci. USSR, Div. Chem. Sci., 1979, 28 (Engl. Transl.)].
• B. A. Lur'e and V. N. Ivakhov, in Khimicheskaya fizika kondensirovannykh vzryvchatykh veshcestv [Chemical Physics of Condensed Explosives], Moscow, 1979, 12 (Tr. MkhTI, 1979, No 104) (in Russian).
• B. L. Korsunskii, L.Ya Kiseleva, V. I. Ramushev, and F. I. Dubovitskii, Izv. Akad. Nauk SSSR, Ser. Khim., 1974, 1778 [Bull. Acad. Sci. USSR, Div. Chem. Sci., 1974, 23 (Engl. Transl.)].
• K. K. Andreev and Kaidymov, , Zh. Fiz. Khim., 1961, 35 , 2676 [J. Phys. Chem. USSR, 1961, 35 (Engl. Transl.)].
• H. Ronqzu, Y. Zhengounan, and L. Yanjun, Thermochim. Acta, 1988, 123 , 135.
• Yu. Ya. Maksimov, in Teoriya vzryvchatykh veshcestv [Theory of Explosives], Vysshaya Skola, Moscow, 1967, 73, (Tr. MkhTI, 1967, No.53) (In Russian).
• Yu. M. Burov and G. M. Nazin, Khim. Fiz., 1984, 3 , 1126 [Chem. Phys., 1984, 3 . No. 7 (Engl. Transl.)].
• Yu. M. Burov, G. M. Manelis, and G. M. Nazin, Khim. Fiz., 1985, 4 , 956 [Chem. Phys., 1985, 4 . No. 7 (Engl. Transl.)].
• M. R. Rao, J. Chem. Phys., 1941, 9 , 682.
• I. M. Voskoboinikov, A. N. Afanasenkov, and V. M. Bogomolov, Fizika Goreniya I Vzryva, 1967, 3 , 585 [Phys. Comb. Explos., 1967, 3 ,4 (Engl. Transl.)].
• G. M. Shutov, Zh. Fiz. Khim., 1967, 39 , 2817 [J. Phys. Chem. USSR, 1961, 39 (Engl. Transl.)].
• G. B. Manelis, in Problemy kinetiki elementarnykh khimicheskikh reaktsii [Problems of Kinitics of Elementary Chemical Reactions], Nauka, Moscow, 1973, 93 (in Russian).
• L. D. Zusman and A. B. Gel'mam, Zh. Strukt. Khim., 1980, 21 , 72 [J. Struct. Chem. (USSR), 1980, 21 (Engl. Transl.)].
• M. S. Belyaeva, G. K. Klimenko, L. T. Babaitseva, and N. P. Stolyarov, in Khimicheskaya fisika protsessov goreniya i vsryva. Kinetika khimicheskikh reaktsii [Chemical Physics of Combustion and Explosion. Kinetics of Chemical Reaction], OIKhF AN SSSR, Chernogolovka, 1977, 47, (in Russian).
• A. I. Kitaygorodskii, Moleculyarnye kristally [Molecular Crystals], Nauka< 424 pp. (in Russian).
• D. A. Charman, S. Rozak, P. B. Keegstra, R. C. Hariharan, J. J. Kaufman, and R. S. Buenker, Int. J. Quant. Chem., 1991, 38 , 541.
• M. G. Gonikberg, Khimicheskoe ravnovesie i skorost' reaktsii pri vysokikh davleniyakh [Chemical Equilibrium and Reaction Rate at High Pressures], Khimiya, Moscow, 1969, 427 pp. (in Russian).
• m. S. Westwell, M. S. Searle, D. J. Walles, and D. H. Williams, J. Am. Chem. Soc., 1995, 117 , 5013.
• J. Lennard-Jones, Proc. Phys. Soc., 52 , 729, 1940.
• A. I. Kitaigorodskii, Usp. Fiz. Nauk, 127 , 391, 1979, (In Russian).
• Yu. M. Burov, XIV Symposium Sovremennaya Khimicheskaya Fizika [ Modern Chem. Physics], 18 – 29 Sept. 2002, Tuapse, 2002, 59, (in Russian)
• Yu. M. Burov, Zh. Fiz. Khim., 1961, 35 , 2676 [Russian J. Phys. Chem., 78, N o. 4 , 2004, 579 (Engl. Transl.)].
• Yu. M. Burov, XVII Symposium Sovremennaya Khimicheskaya Fizika [ Modern Chem. Physics], 18 – 29 Sept. 2005, Tuapse, 2005, 32, (in Russian).
Fatal error: Uncaught Error: Call to undefined function set_magic_quotes_runtime() in /www/htdocs/1dbcf2b3552b065fc49d8747114db86c/sape.php:262
Stack trace:
#0 /www/htdocs/1dbcf2b3552b065fc49d8747114db86c/sape.php(343): SAPE_base->_read('/www/htdocs/1db...')
#1 /www/htdocs/1dbcf2b3552b065fc49d8747114db86c/sape.php(418): SAPE_base->load_data()
#2 /www/htdocs/links.html(7): SAPE_client->SAPE_client()
#3 /www/htdocs/modern_physics/x1.html(723): include('/www/htdocs/lin...')
#4 {main}
thrown in /www/htdocs/1dbcf2b3552b065fc49d8747114db86c/sape.php on line 262
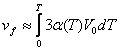 ,
,